|
|
|
Project:
Rephetio: Repurposing drugs on a hetnet [rephetio]
Publication:
Evolutionary Signatures amongst Disease Genes Permit Novel Methods for Gene Prioritization and Construction of Informative Gene-Based Networks
|
Status:
Completed
Views
103
Topics
Referenced by
Cite this as
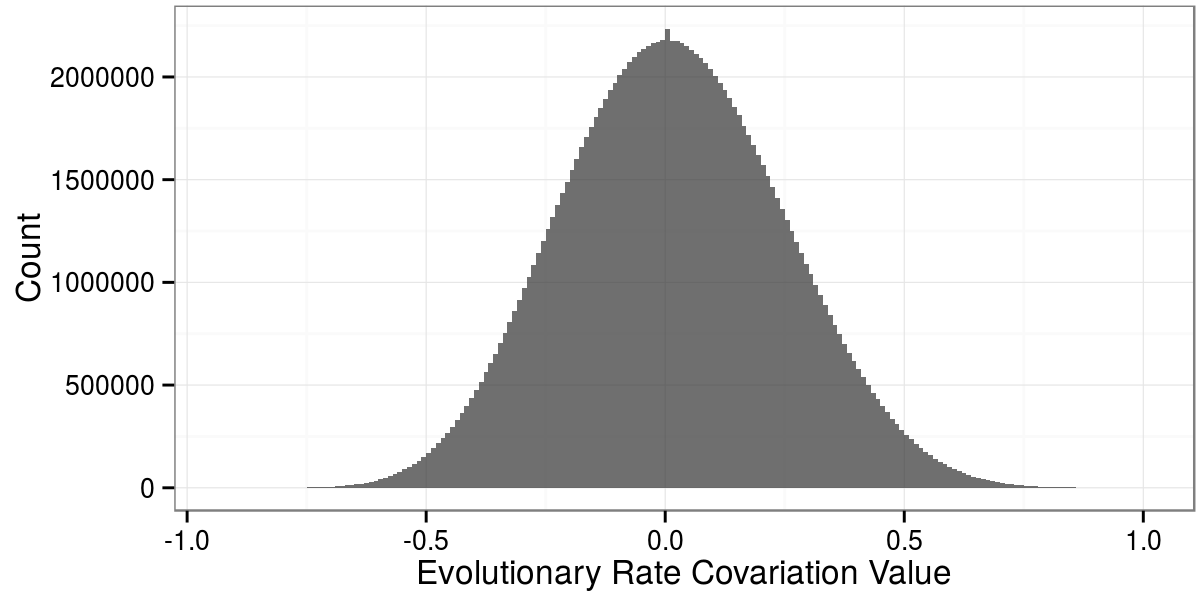
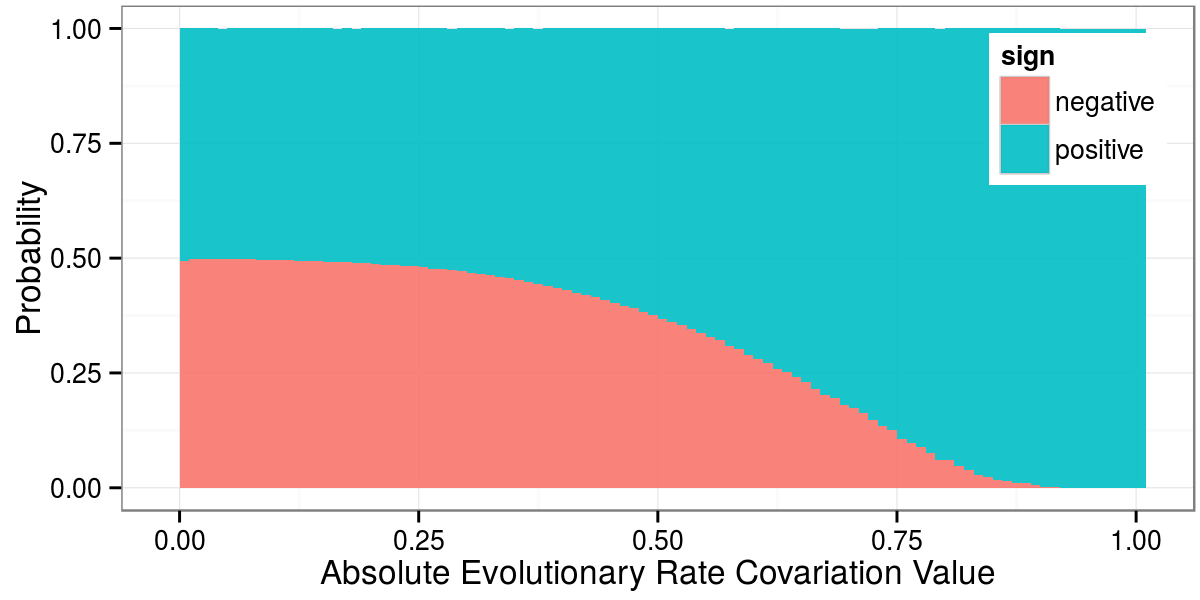
Daniel Himmelstein, Raghavendran Partha (2015) Selecting informative ERC (evolutionary rate covariation) values between genes. Thinklab. doi:10.15363/thinklab.d57
License
Share
|