|
|
|
Project:
Rephetio: Repurposing drugs on a hetnet [rephetio]
Publication:
DrugBank 4.0: shedding new light on drug metabolism
|
Status:
Completed
Views
994
Topics
Referenced by
Cite this as
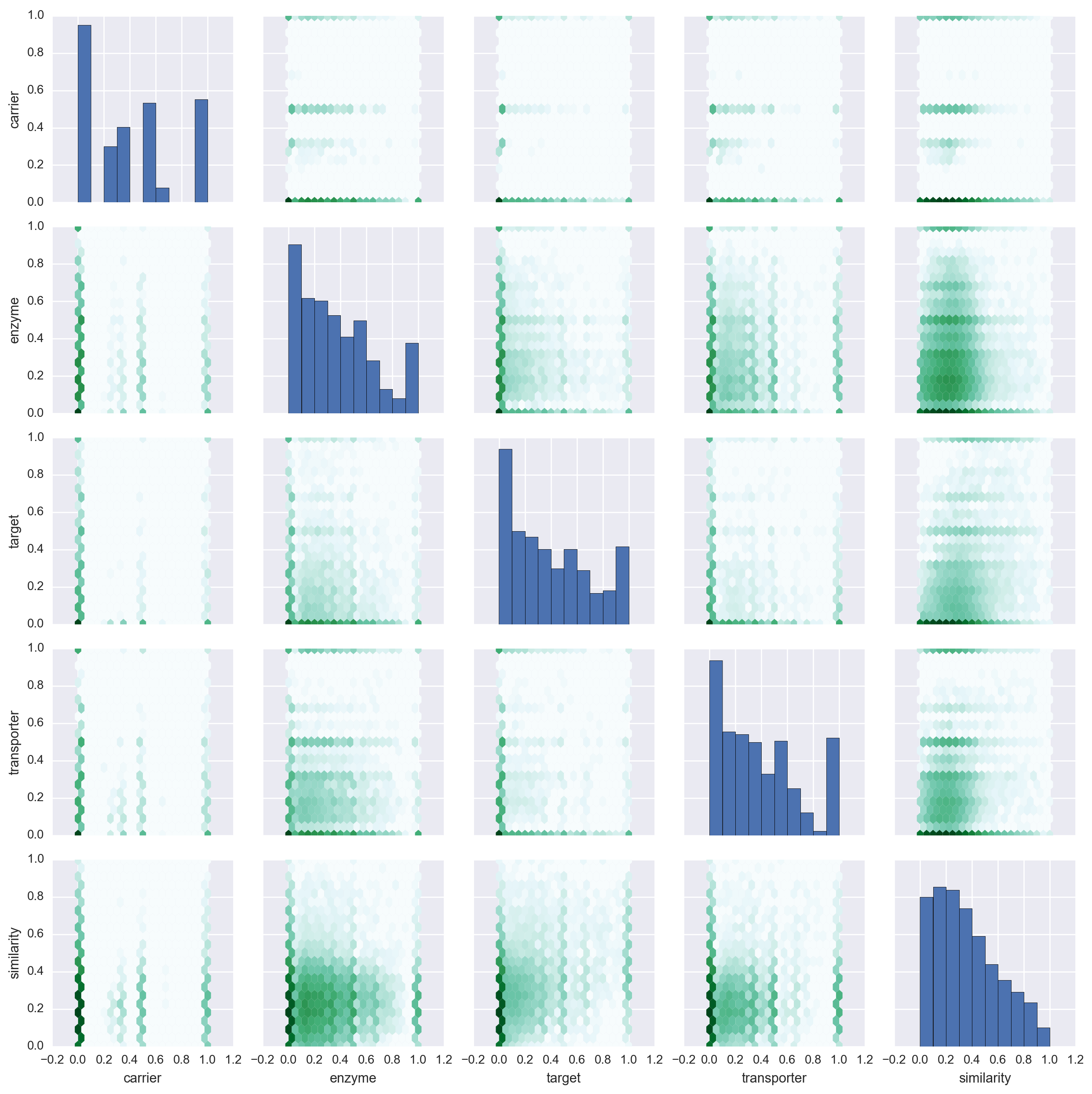
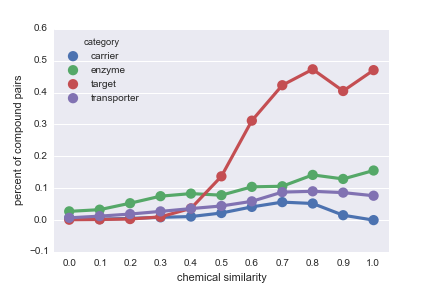
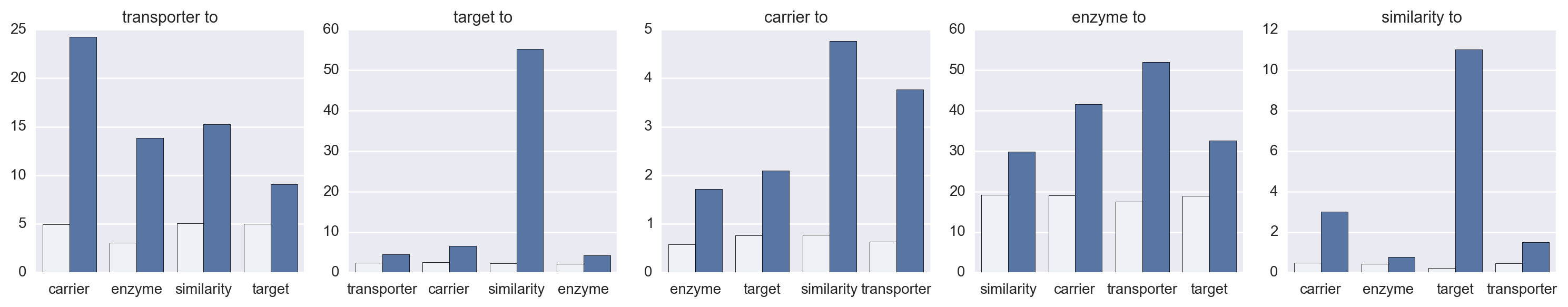
Daniel Himmelstein, Sabrina Chen (2015) Protein (target, carrier, transporter, and enzyme) interactions in DrugBank. Thinklab. doi:10.15363/thinklab.d65
License
Share
|



{kind=link}